Nasljedna multipla egzostoza
Nasljedni multipli osteohondromi (HMO), također poznati kao nasljedne multiple egzostoze, je poremećaj karakteriziran razvojem višestrukih benignih osteohrskavičnih masa (egzostoza) u odnosu na krajeve dugih kostiju donjih udova, kao što su butne kosti i tibije i gornjih udova kao što su kosti nadlaktica i podlaktica. Također su poznati kao osteohondromi. Dodatna mjesta pojave uključuju ravne kosti kao što su karlična kost i lopatica. Raspodjela i broj ovih egzostoza pokazuju veliku raznolikost među oboljelim osobama. Egzostoze se obično javljaju u djetinjstvu. Ogromna većina oboljelih osoba se klinički manifestira do trenutka kada dostignu adolescenciju.[1][2] Mali postotak oboljelih osoba je u opasnosti od razvoja maligne transformacije, odnosno sarkome. Incidencija nasljednih višestrukih egzostoza je oko 1 na 50.000 osoba.[3] Nasljedni multipli osteohondromi je preferirani termin koji koristi Svjetska zdravstvena organizacija.
| Nasljedni multipli osteohondromi (Nasljedne multiple egzostoze) | |
|---|---|
 Fotografija nogu 26-godišnjeg muškarca, sa više kvržica koje dovode do deformacija. | |
| Klasifikacija i vanjski resursi | |
| ICD-10 | Q78.6 |
| ICD-9 | 756.59 |
| OMIM | 133700 OMIM: 133701 |
| DiseasesDB | 33342 |
| MeSH | D005097 |
Prezentacija
urediPrimjetna kvržica na nekom od ekstremiteta može biti prvi simptom. Mogu nastati višestruke deformacije, odnosno deformacije koronske ravni oko koljena, gležnjeva, ramena, laktova i zapešća. Naprimjer, susreću se genu valgum (koljena), valgus skočnog zgloba, savijanje i skraćivanje ulni, te radijalna subluksacija glave. Većina oboljelih osoba ima klinički manifestne osteohondroma oko koljena. Uključenost podlaktice u HMO je značajna.[1][4] Nadalje, može doći do niskog rasta i općenito je nesrazmjeran. Takve manifestacije obično su posljedica poremećaja fize rasta, posebno što se osteohondromi obično javljaju na metafiznim krajevima dugih kostiju u neposrednoj blizini tijela.[1][4] Unutarzglobni osteohondromi kuka mogu induciraju ograničenje opsega pokreta, bol u zglobovima i acetabulumsku displaziju.[2] Isto tako, može doći do bolova u zglobovima na drugim lokacijama i neurovaskularne kompresije. Nadalje, funkcionalna onesposobljenost u pogledu aktivnosti svakodnevnog života može biti prisutna karakteristika. Bol zbog deformacije kičme ili neurološki kompromis bi trebali izazvati sumnju na zahvaćenost pršljenova.[3]
Moguća povezanost sa autizmom
urediNeki roditelji djece sa HME primijetili su socijalne probleme nalik autizmu kod svoje djece. Da bi se ta zapažanja dublje istražila, studija iz 2012. koju je sproveo Sanford-Burnham Medical Research Institute koristila je mišji model HME za promatranje kognitivnih funkcija. Nalazi su pokazali da mutantni miševi ispoljavaju tri autistične karakteristike: socijalno oštećenje, oštećenje ultrazvučne vokalizacije i ponašanje koje se ponavlja.[5]
Genetika
urediHME je autosomno dominantni nasljedni poremećaj. To znači da pacijent sa HME ima 50% šanse da ovaj poremećaj prenese na svoju djecu. Većina osoba sa HME ima roditelja koji takođe ima to stanje, međutim, otprilike 10%–20% osoba sa HME ima bolest kao posljedicu spontane mutacije i stoga su prva osoba u svojoj porodici koja je pogođena.
HME je do sada bio povezan s mutacijama u tri gena:
- EXT1 koji se nalazi na hromosomskom segmentu 8q24.1[6]
- EXT2 na 11p13[7] i
- EXT3 na krtatkom (p) krtaku hromosoma 19 (iako smatrase da njegova lokacija nije precizno utvrđena)[8]
Mutacije u ovim genima obično dovode do sinteze skraćenog EXT proteina koji ne funkcionira normalno. Poznato je da su EXT proteini važni enzimi u sintezi heparan-sulfat; međutim, nejasan je tačan mehanizam kojim je izmijenjena sinteza heparan-sulfata koja bi mogla dovesti do abnormalnog rasta kostiju povezanog s HME. Smatra se da može uticati na normalnu proliferaciju hondrocita i ćelijsku diferencijaciju, što dovodi do abnormalnog rasta kostiju.[9][10] Budući da su geni HME uključeni u sintezu glikana (heparan-sulfat), HME se može smatrati kongenitalnim poremećajem glikozilacije prema novoj nomenklaturi CDG, predloženoj u 2009.[11]
Za osobe sa HME koje razmišljaju o osnivanju porodice, dostupni su preimplantacijsko genetičko testiranje i prenatalna dijagnoza, kako bi se utvrdilo da li je njihovo nerođeno dijete naslijedilo bolest. HME ima penetrabilnost od 96%, što znači da ako se zahvaćeni gen zaista prenese na dijete, ono će imati 96% stvarnog manifestiranja bolesti i 4% šanse da ima bolest, ali je nikada neće manifestirati. Iznos penetracije od 96% dolazi iz samo jedne studije.[12] Druge studije su uočile i nepotpunu i promjenjivu penetraciju, ali bez izračunavanja postotka penetrabilnosti, npr.[13] U obje gore spomenute studije, osobe bez simptoma koje su nosile neispravan gen bile su pretežno žene, što je dovelo do nagađanja da je nepotpuna penetrabilčnost vjerojatnije biti izražena kod žena. Zaista, drugi radovi su pokazali da dječaci/muškarci imaju teže bolesti od žena, kao i da broj egzostoza kod oboljelih članova iste porodice može značajno varirati.[14]
Veća je vjerovatnoća da će simptomi biti ozbiljni ako je mutacija na genu ext1 umjesto ext2 ili ext3; "ext1" je također najčešće zahvaćeni gen kod pacijenata sa ovim poremećajem.[14]
Patofiziologija
urediOvaj sindrom karakterizira rast benignih tumora kostiju pokrivenih hrskavicom oko područja aktivnog rasta kosti, posebno metafiza dugih kostiju. Obično se pet ili šest egzostoza nalazi u gornjim i donjim udovima. Najčešće lokacije su:[15]
HME može dovesti do skraćivanja i savijanja kostiju; pogođene osobe često imaju nizak rast. Ovisno o lokaciji, egzostoze mogu uzrokovati sljedeće probleme: bol ili utrnulost zbog kompresije živca, vaskularni kompromis, nejednakost dužine udova, iritacija tetiva i mišića, Madelungova deformacija[16] kao i ograničeni opseg pokretljivosti u zglobovima na koje zadiru. Kao odrasla osoba, pogođeni ima povećan rizik od razvoja rijetkog oblika raka kostiju zvanog hondrosarkom.[16] Problemi se mogu pojaviti u kasnijem životu, a oni mogu uključivati slabe kosti i oštećenje živaca.[17][18][19] Prijavljena stopa transformacije kreće se od čak 0,57%[12] čak 8,3% ljudi sa HME.[20]
Dijagnoza
urediDijagnoza HMO zasniva se na uspostavljanju tačnih korelacija između gore navedenih kliničkih obilježja i karakterističnih radiografskih svojstava. Porodična anamneza može dati važan trag za dijagnozu. Ovo je dopunjeno testiranjem za dva gena za koje je poznato da patogene varijante uzrokuju HMO: EXT1 i EXT2. Kombinacija analize sekvence i analize delecije čitavih kodirajućih regija i EXT1 i EXT2 otkriva patogene varijante kod 70–95% oboljelih osoba.[3][4] Obilježje adiografske dijagnoze je prisustvo osteohondroma na metafiznim krajevima dugih kostiju u kojima korteks i medula osteohondroma predstavljaju kontinuirani produžetak kosti domaćina. Ovo se lahko može dokazati na rendgenskim snimcima koljena.[1][3]
Liječenje
urediIndikacije za hiruršku intervenciju kod osoba sa HMO ostaju nejasne i uveliko variraju u medicinskoj literaturi. Općenito, hirurško liječenje HMO uključuje jedan ili više od sljedećih postupaka: eksciziju ostehondroma, postepeno ili akutno produženje kosti kao što je produženje lakatne kosti, korektivne osteotomije, privremena hemiepifiziodeza za ispravljanje deformacija ugaonih zglobova kao što su hemiepifiziodeza distalnog radijusa i hemiepifiziodeza distalnog poluprečnika i medijalna distalna tibiodeza.[1][3] Ipak, malo je dokaza koji podržavaju tekuću pedijatrijskoortopedsku praksu kod nasljednih multiplih osteohondroma. Nedavni sistematski pregledi otkrili su nedovoljne dokaze koji bi dokazali da tekuće hirurško liječenje HMO značajno poboljšava funkciju ili da dokaže da utiče na kvalitet života pogođene djece.[1][2] Da bi se povećala količina dokaza u medicinskoj literaturi, date su određene preporuke. Izgradnja dobro osmišljenih prospektivnih studija koje mogu pružiti jasniji odnos između hirurških zahvata, karakteristika pacijenata i ishoda je velika potražnja. U suprotnom, praćenje trenutnog dizajna studije nastavit će postavljati više pitanja nego odgovora.[1][2] Totalna artroplastika kuka korištena je za liječenje teške i bolne HMO kukovog zgloba. Totalna artroplastika kuka kod osoba sa HMO je izazovna zbog distorzije anatomije i ponovljenih operacija koje se izvode radi rješavanja pritužbi povezanih s egzostozom.[21]
Epidemiologija
urediProcjenjuje se da se HME javlja kod 1 od 50.000 ljudi.[15][19]
Dodatne slike
uredi-
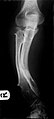 Mujltipli osteohondromi koji uzrokuju deformacije podlaktice (skraćivanje radijusa sa sekundarnim savijanjem ulne).
Mujltipli osteohondromi koji uzrokuju deformacije podlaktice (skraćivanje radijusa sa sekundarnim savijanjem ulne). -
 Multipli osteohondromi na karlici
Multipli osteohondromi na karlici -
 Multipli osteohondromi oko koljena
Multipli osteohondromi oko koljena -
 CT-snimak osteohondroma u MO
CT-snimak osteohondroma u MO
.jpg)
.jpg)


Reference
uredi- ^ a b c d e f g EL-Sobky, TA; Samir, S; Atiyya, AN; Mahmoud, S; Aly, AS; Soliman, R (21. 3. 2018). "Current paediatric orthopaedic practice in hereditary multiple osteochondromas of the forearm: a systematic review". Sicot-J. 4: 10. doi:10.1051/sicotj/2018002. PMC 5863686. PMID 29565244.
- ^ a b c d Makhdom, AM; Jiang, F; Hamdy, RC; Benaroch, TE; Lavigne, M; Saran, N (20. 5. 2014). "Hip joint osteochondroma: systematic review of the literature and report of three further cases". Advances in Orthopedics. 2014: 180254. doi:10.1155/2014/180254. PMC 4054980. PMID 24963411.
- ^ a b c d e Wuyts, W; Schmale, GA; Chansky, HA; et al. (21. 11. 2013). "Hereditary Multiple Osteochondromas". GeneReviews. University of Washington, Seattle. Pristupljeno 24. 3. 2018.
- ^ a b c Alvarez, CM; De Vera, MA; Heslip, TR; Casey, B (septembar 2007). "Evaluation of the anatomic burden of patients with hereditary multiple exostoses". Clin Orthop Relat Res. 462: 73–79. doi:10.1097/BLO.0b013e3181334b51. PMID 17589361. S2CID 39999620.
- ^ Irie F, Badie-Mahdavi H, Yamaguchi Y (mart 2012). "Autism-like socio-communicative deficits and stereotypies in mice lacking heparan sulfate". Proceedings of the National Academy of Sciences of the United States of America. 109 (13): 5052–6. Bibcode:2012PNAS..109.5052I. doi:10.1073/pnas.1117881109. PMC 3323986. PMID 22411800.
- ^ Cook A, Raskind W, Blanton SH, Pauli RM, Gregg RG, Francomano CA, Puffenberger E, Conrad EU, Schmale G, Schellenberg G (1993). "Genetic heterogeneity in families with hereditary multiple exostoses". American Journal of Human Genetics. 53 (1): 71–9. PMC 1682231. PMID 8317501.
- ^ Wu YQ, Heutink P, de Vries BB, Sandkuijl LA, van den Ouweland AM, Niermeijer MF, Galjaard H, Reyniers E, Willems PJ, Halley DJ (1994). "Assignment of a second locus for multiple exostoses to the pericentromeric region of chromosome 11". Human Molecular Genetics. 3 (1): 167–71. doi:10.1093/hmg/3.1.167. PMID 8162019.
- ^ Le Merrer M, Legeai-Mallet L, Jeannin PM, Horsthemke B, Schinzel A, Plauchu H, Toutain A, Achard F, Munnich A, Maroteaux P (1994). "A gene for hereditary multiple exostoses maps to chromosome 19p". Human Molecular Genetics. 3 (5): 717–22. doi:10.1093/hmg/3.5.717. PMID 8081357.
- ^ Zak BM, Crawford BE, Esko JD (2002). "Hereditary multiple exostoses and heparan sulfate polymerization". Biochimica et Biophysica Acta (BBA) - General Subjects. 1573 (3): 346–55. doi:10.1016/S0304-4165(02)00402-6. PMID 12417417.
- ^ Stieber JR, Dormans JP (2005). "Manifestations of hereditary multiple exostoses". The Journal of the American Academy of Orthopaedic Surgeons. 13 (2): 110–20. doi:10.5435/00124635-200503000-00004. PMID 15850368. S2CID 29077708.
- ^ Jaeken J, Hennet T, Matthijs G, Freeze HH (2009). "CDG nomenclature: time for a change!". Biochim. Biophys. Acta. 1792 (9): 825–6. doi:10.1016/j.bbadis.2009.08.005. PMC 3917312. PMID 19765534.
- ^ a b Legeai-Mallet L, Munnich A, Maroteaux P, Le Merrer M (juli 1997). "Incomplete penetrance and expressivity skewing in hereditary multiple exostoses". Clinical Genetics. 52 (1): 12–6. doi:10.1111/j.1399-0004.1997.tb02508.x. PMID 9272707. S2CID 44423092.
- ^ Faiyaz-Ul-Haque M, Ahmad W, Zaidi SH, et al. (august 2004). "Novel mutations in the EXT1 gene in two consanguineous families affected with multiple hereditary exostoses (familial osteochondromatosis)". Clinical Genetics. 66 (2): 144–51. doi:10.1111/j.1399-0004.2004.00275.x. PMID 15253765. S2CID 10431219.
- ^ a b Porter DE, Lonie L, Fraser M, et al. (septembar 2004). "Severity of disease and risk of malignant change in hereditary multiple exostoses. A genotype-phenotype study". The Journal of Bone and Joint Surgery. British Volume. 86 (7): 1041–6. doi:10.1302/0301-620x.86b7.14815. hdl:20.500.11820/8754788e-ceea-4613-8e65-60d34fcf9edc. PMID 15446535.
- ^ a b Turek's orthopaedics principles and their application (6th izd.). Philadelphia: Lippincott Williams & Wilkins. 2005. str. 263. ISBN 9780781742986.
- ^ a b Davies, A. Mark; Pettersson, Holger (2002). Pettersson, Holger; Ostensen, Harald (ured.). Radiography of the Musculoskeletal System (PDF). Geneva: World Health Organization. str. 177, 189. ISBN 978-92-4-154555-6. Arhivirano s originala (PDF), 27. 11. 2014. Pristupljeno 17. 1. 2022.
- ^ CANNON JF (1954). "Hereditary multiple exostoses". American Journal of Human Genetics. 6 (4): 419–25. PMC 1716573. PMID 14349947.
- ^ McBride WZ (septembar 1988). "Hereditary multiple exostoses". American Family Physician. 38 (3): 191–2. PMID 3046271.
- ^ a b Schmale GA, Conrad EU, Raskind WH (juli 1994). "The natural history of hereditary multiple exostoses". The Journal of Bone and Joint Surgery. American Volume. 76 (7): 986–92. doi:10.2106/00004623-199407000-00005. PMID 8027127. Arhivirano s originala, 20. 9. 2014.
- ^ Kivioja A, Ervasti H, Kinnunen J, Kaitila I, Wolf M, Böhling T (mart 2000). "Chondrosarcoma in a family with multiple hereditary exostoses". The Journal of Bone and Joint Surgery. British Volume. 82 (2): 261–6. doi:10.1302/0301-620X.82B2.0820261. PMID 10755438.
- ^ Vaishya, R; Swami, S; Vijay, V; Vaish, A (5 Jan 2015). "Bilateral total hip arthroplasty in a young man with hereditary multiple exostoses". BMJ Case Rep. 2015: bcr2014207853. doi:10.1136/bcr-2014-207853. PMC 4289752. PMID 25564594.
Vanjski linkovi
uredi